
Magic-BLAST version 1.5.0 is here~
иҪҜ件еҢ…зҡ„дёӢиҪҪең°еқҖ:
https://ftp.ncbi.nlm.nih.gov/blast/executables/magicblast/LATEST/
иӢҘдҪ жӣҫз»ҸжңүдёӘжғіжі•пјҢжғіе…·дҪ“ең°йҮҚж–°з»ҳеҲ¶еҮәдёҖе№…жӣҙеӨ§зҡ„е…Ёеҹәеӣ з»„RNAжҲ–DNAеәҸеҲ—еӣҫпјҢд»ҘеҫҖйғҪиҰҒз”Ёblastи§ЈеҶіпјҢиҖҢзҺ°еңЁдҪ еҸҜд»Ҙз”Ёmagic-blastе·Ҙе…·е®һзҺ°дәҶгҖӮ
е°Ҹзј–д»ҠеӨ©д»Ӣз»ҚиҝҷзҜҮдәҺ2019е№ҙ7жңҲеҸ‘иЎЁеңЁBMC Bioinformaticsзҡ„ж–Үз« гҖӮ

Magic-BLASTе·Ҙе…·йҖҡз”ЁжҖ§йқһеёёејәеӨ§пјҢиҝҗиЎҢд№ҹеҫҲеҝ«гҖӮе®ғеҸҜе°ҶиҰҒиҜ»еҸ–зҡ„зүҮж®өдёҺBLASTж•°жҚ®еә“жҲ–дёҖдёӘFASTAж–Ү件жҺ’еҲ—еңЁдёҖжҺ’гҖӮеҸҜжҺҘеҸ—FASTQж–Ү件дҪңдёәиҫ“е…ҘпјҢжҲ–иҖ…иҮӘеҠЁд»ҺNCBIзҡ„SRAеӯҳеӮЁеә“дёӯжЈҖзҙўгҖӮ
йӮЈдҪ зҹҘйҒ“Magic-BLASTз®—жі•жҖҺж ·еҒҡзҡ„еҗ—пјҹжқҘзңӢзңӢиҝҷжөҒзЁӢеӣҫвҖҰвҖҰ
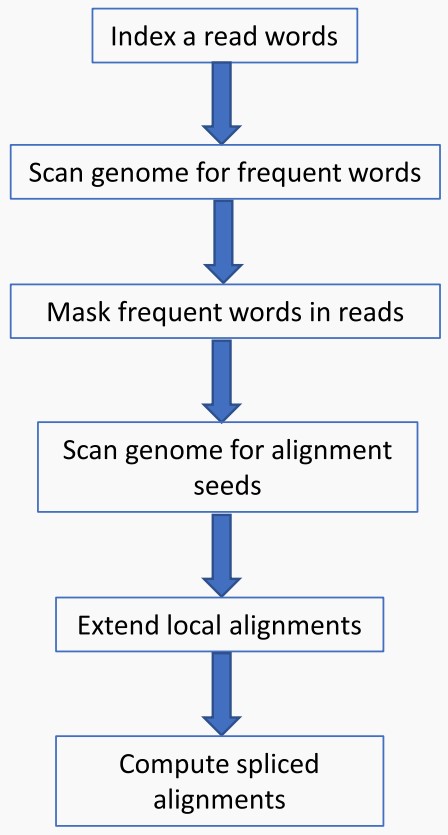
з»ҷдҪ зңӢдёҖдёӘдҫӢеӯҗе°ұжҳҺзҷҪдәҶгҖӮ

дҪҝз”Ёж–№жі•пјҡ
1пјҺ жһ„е»әж•°жҚ®еә“
1 makeblastdb -in Malus_baccata.fasta -dbtype nucl -parse_seqids -out Malus_baccata
-inГ еҸӮиҖғеәҸеҲ—
-dbtypeГ ж•°жҚ®зұ»еһӢпјҡж ёиӢ·й…ёе’ҢиӣӢзҷҪиҙЁеҸҜйҖү
-parse_seqidsГ жҡӮж—¶иҝҳжІЎжҗһжҮӮиҝҷдёӘеҸӮж•°зҡ„ж„ҸжҖқ
-outГ ж•°жҚ®еә“зҡ„еҗҚз§°
2пјҺ жҜ”еҜ№еәҸеҲ—
# й»ҳи®Өиҫ“е…Ҙж–Ү件дёәfastaж јејҸ# еҚ•дёӘfastaж–Ү件magicblast -query reads.fasta -db Malus_baccata# дёӨдёӘfastaж–Ү件magicblast -query reads.fasta -query_mate mates.fasta -db Malus_baccata# еҰӮжһңиҫ“е…Ҙж–Ү件дёәfastqж јејҸmagicblast -query reads.fastq -db Malus_baccata -infmt fastq# еҸҢз«Ҝж•°жҚ®magicblast -query reads_R1.fastq -query_mate reads_R2.fastq -db Malus_baccata вҖ“infmt fastq
3пјҺ еҸҜеҸҳеүӘеҲҮ
By default, Magic-BLAST aligns RNA reads to a genome and reports spliced alignmets.
4пјҺ еӨҡзәҝжҖ§еҸӮж•°
magicblast -query reads.fasta -db genome -num_threads 10
еҸӮиҖғж–ҮзҢ®пјҡ
Grzegorz M. Boratyn, Jean Thierry-Mieg, Danielle Thierry-Mieg, Ben Busby and Thomas L. Madden. Magic-BLAST, an accurate RNA-seq aligner
for long and short reads. BMC Bioinformatics (2019) 20:405
вҖў END вҖў

